
三峽大學李東升組《CEJ》:構筑新型、高效、耐用的助催化劑用于光催化產氫
2024-04-23 13:09:07
作者:材料科學與工程 來源:材料科學與工程
分享至:


第一作者:喬秀清;通訊作者:蘭亞乾、李東升
通訊單位:華南師范大學、三峽大學
論文DOI:https://doi.org/10.1016/j.cej.2022.140791

全文速覽
助催化劑輔助的光催化制氫可以明顯的提高光催化活性,但材料的穩定性較差。本文提出通過調節助催化劑的組成和結構來構筑高效、穩定的光催化助劑,利用原位碳化策略準確合成了一種奇妙的助催化劑,即強錨定在導電碳基質中的MoO2/Mo2C-C納米顆粒,并將其用作CdS光催化析氫的高效、穩健的助催化劑。令人驚喜的是,所獲得的MoO2/Mo2C-C助催化劑非常穩定,在大氣條件下儲存兩年也能保持原始活性。優化的MoO2/Mo2C-CdS-0.3(MMCC-0.3)光催化劑,在無需補充二次犧牲劑的條件下,產氫活性為18.43 mmol h−1 g−1,展示出連續90小時測試(在15天內進行)的超穩定性。實驗結果和DFT計算都表明,獨特的MoO2/Mo2C-C助催化劑不僅具有最佳的氫結合能(ΔG*)、降低的d帶中心及析氫反應動力學勢壘,導電碳橋、高質量界面和暴露出豐富的CdS活性位點,協同作用增強了可見光吸收,促進了電荷分離,保證了MoO2/Mo2C-C-CdS光催化劑的優異活性和穩定性。這項工作不僅為實際應用提供了一種高效、低成本的助催化劑,而且為設計用于光催化劑及其他用途的堅固助催化劑提供了寶貴的見解。
背景介紹
隨著全球工業化進程,人類對清潔能源的需求越來越大,光催化分解水產氫可利用太陽能和水制備氫能,滿足可持續發展的要求,是一種極具前景的策略。為進一步提升光催化劑產氫活性,助催化劑修飾策略引起了人們關注。發展高活性、高穩定性非貴金屬助催化劑對促進光催化產業化進程極具研究意義。
具有特殊電子構型的鉬基材料在光/電催化劑中越來越受到關注,其費米能級(Ef)位于Mo元素的4d軌道內,氧化鉬(MoO2)在電導率(~6×103S m-1)和金屬性質方面表現出無與倫比的優勢。然而,MoO2多通過高溫煅燒工藝制取,顆粒易于團聚、活性位點暴露困難,對催化性能不利。此外,MoO2與H中間物種的結合能力較差,導致產氫動力學緩慢,設計和探索具有良好分散性、豐富暴露活性位點和最佳電子結構的高效MoO2助催化劑,對于構建高效的助催化劑具有重要意義。
本文亮點
1.采用前驅體熱解及原位碳化工藝獲得了一種高效、超穩定的MoO2/Mo2-C助催化劑。
2.經過優化的MMCC光催化劑具有顯著提高光催化的活性,超過90小時的穩定性。
3.該工作首次通過助催化劑組分和結構優化,實現了d帶中心調控,提升助催化劑的活性及穩定性。
4.該助催化劑綜合性能優于貴金屬Pt,在實際應用中顯示出良好的前景。
圖文解析
通過苯胺離子C6H8N+和鉬酸根離子Mo3O102-在堿性條件下的靜電組裝生成Mo3O10(C6H8N)2·2H2O前驅體,以前驅體為自犧牲模板通過改變熱處理溫度得到具有不同組分和結構的助催化劑。結合熱重、XRD確定了助催化劑的生成過程為:

顯然,通過MoO2的部分原位碳化可以獲得錨定在碳基體上的MoO2/Mo2C-C雜化物(MoO2/Mo2C-C),這有望在組合物之間形成高質量的界面。然后,通過超聲混合MoO2/Mo2C-C助催化劑和所制備的CdS-NRs,獲得了MoO2/MO2-C-C-CdS(MMCC)光催化劑。
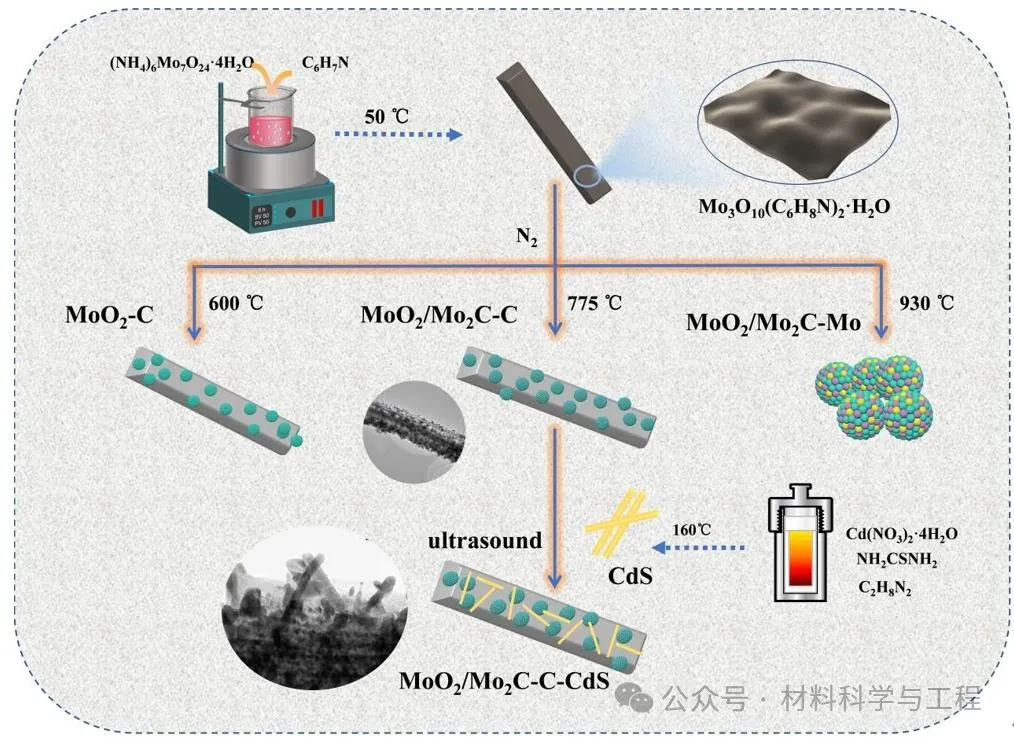
圖1樣品的制備過程示意圖
XRD圖譜和拉曼光譜證實了600°C、775°C和930°C下獲得的產物分別為MoO2-C、MoO2/Mo2C-C和MoO2/Mo2C-Mo,單斜MoO2(JCPDS No.32-0671)和立方Mo2C(JCPDSs No.15-0457)及碳物種被觀察到。SEM和TEM顯示結晶、分散良好的MoO2/Mo2C-NP錨定在無定型的碳骨架上。圖2g的間距為0.342nm和0.207nm的清晰晶格條紋分別歸屬于MoO2和Mo2C的(200)面。導電碳不僅防止助催化劑的聚集,還為電荷載流子的傳輸提供了“高速公路”,這將有利于電荷載流子的分離。

圖2 MoO2/Mo2C-C的結構表征
應用Mo K邊緣的X射線吸收結構(XAS)揭示了MoO2-C和MoO2/Mo2C中Mo物種的化學狀態和局部配位環境。X射線近邊緣吸收結構(XANES)表明,由于較高的氧化態,MoO2-C具有最高的能量吸收邊緣(E0)。MoO2/Mo2C-C樣品中Mo物種的E0位于Mo箔和MoO2C的E0之間,表明Mo帶正電,平均化學價在Mo0和Mo4+之間。MoO2/Mo2C-C中Mo K邊緣的總體特征與MoO2-C的特征非常相似,揭示了相似的配位結構。擴展XAFS(FT-EXAFS)曲線的相應傅立葉變換(FT)k3加權χ(k)函數表明,Mo箔在2.36Å處的峰值(未經相位校正)源于Mo-Mo反向散射配位。根據MoO2-C樣品,在1.56和3.34Å處的兩個主峰可分別歸屬于第一和第二配位殼中的Mo-O和Mo–Mo。2.17Å處的弱峰源于Mo-Mo的背散射。至于MoO2/Mo2C-C,1.56Å的峰可以與Mo-C/O的貢獻相擬合,這很難區分,因為Mo–O和Mo–C的鍵長相似。然而,MoO2/Mo2C-C中較短的Mo–Mo鍵是由摻雜到MoO2中的C原子引起的。此外,與MoO2-C相比,由于Mo-C配位的散射貢獻,該峰的振幅大大增加。2.14Å(Mo–Mo)和3.19Å(Mo-Mo)處的分辨良好的峰顯示出與MoO2-C相似的散射路徑。通過在R空間中對FT-EXAFS光譜的定量擬合,確定了Mo散射中心周圍的C/O原子的配位數為2.00±0.34(圖3c-d,表S1)。Mo-C/O殼層的平均距離為2.00±0.03Å。分析了樣品Mo K邊緣XANES的一階導數,以揭示Mo物種的電子密度,圖如圖所示。第3e段。觀察到的MoO2/Mo2C-C(20003.1 eV)的最大值位于Mo箔(20000.0 eV)和MoO2-C(20008.8 eV)之間,進一步表明Mo物種的氧化狀態。圖3f展示了Mo K邊的半能和價態之間的關系。通過線性擬合,Mo在MoO2/Mo2C-C中的平均氧化態估計為+1.75,表明Mo處于缺電子狀態。基于Morlet小波對Mo K邊EXAFS振蕩進行小波變換EXAFS(WT-EXAFS),以揭示R和K空間中的徑向距離分辨率,如圖3g所示,Mo箔在~8.2Å−1處的強度最大值可以歸屬于Mo-Mo的貢獻。MoO2-C在~4.4和~12.3Å−11處的強度最大值歸屬于Mo-O和Mo-Mo的貢獻。對于MoO2/Mo2C-C,在~6.5Å−1處的WT最大中心對應于Mo-C/O,而在~11Å−1的另一個最大中心與Mo-Mo貢獻相關,證明了樣品中MoO2和Mo2C的形成。
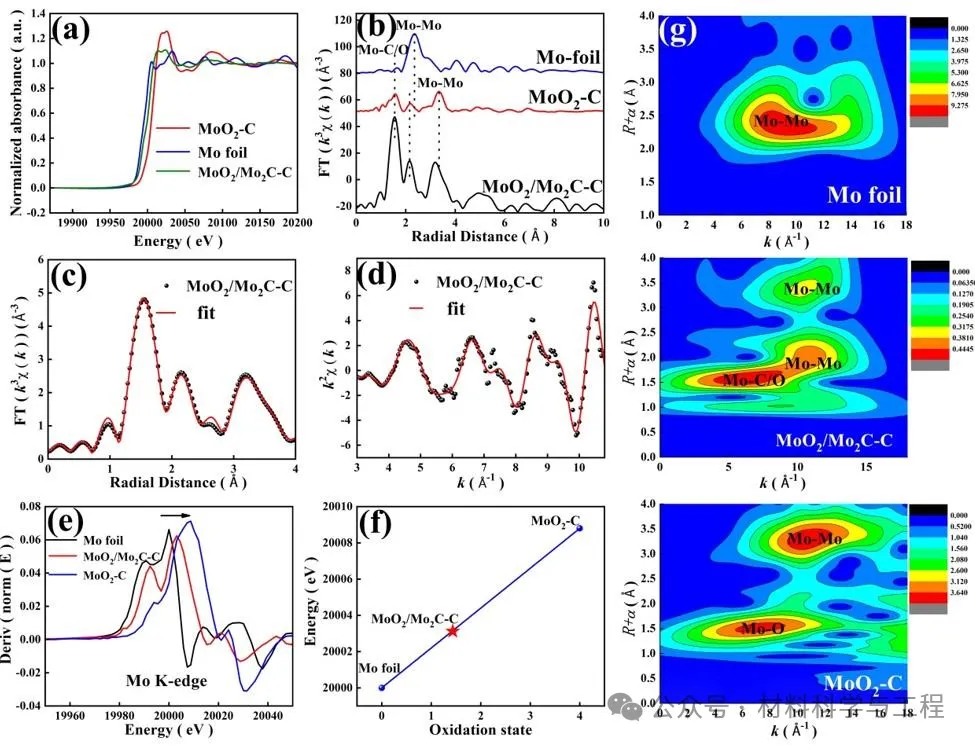
圖3 MoO2/Mo2C-C的表征
為了評估MoO2/Mo2C-C助催化劑的活性,評估了MMCC樣品的光催化析氫活性,并對MoO2-C、MoO2/Mo2C-Mo、C和商業鉑碳改性的CdS進行了比較研究。單獨的CdS表現出3.78mmol h-1g-1的低析氫速率,而MoO2/Mo2C-C助催化劑沒有檢測到析氫。當MoO2/Mo2-C與CdS結合時,析氫速率顯著提高,表明MoO2/MO2-C僅作為光催化析氫的助催化劑。在MMCC光催化劑中,MMCC-0.3樣品提供了18.43 mmol h-1 g-1的最高H2產生率,這比裸CdS提高了4.87倍。值得注意的是,MMCC-0.3表現出比其他助催化劑改性的CdS高得多的光催化活性。耐久性測試表明在沒有補充二次犧牲劑的情況下,MMCC-0.3在連續90小時的長期耐久性測量(在15天內進行)中顯示出相同的析氫活性,表明其具有優異的穩定性。令人興奮的是,即使在大氣條件下儲存2年,MoO2/Mo2C-C助催化劑的活性也能很好地保持。相反,對照樣品的析氫速率在18小時內嚴重惡化。因此,Mo2C不僅可以提高MoO2的助催化活性,而且具有超長期的穩定性,這對實際應用至關重要。
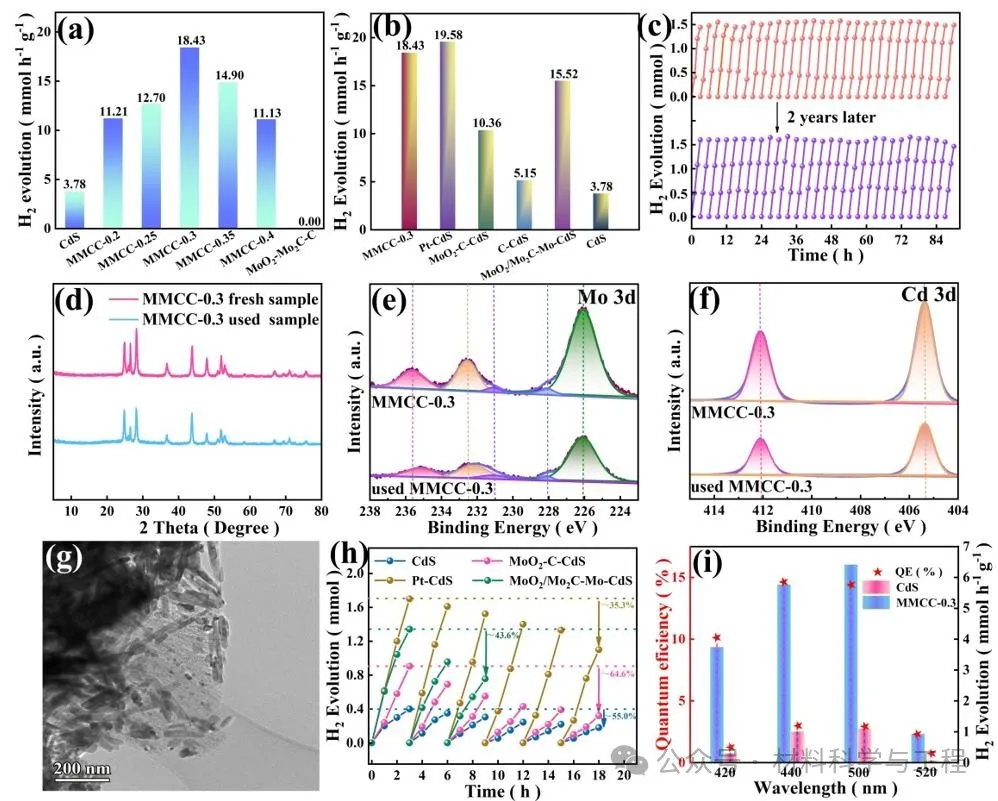
圖4 MoO2/MO2-C-C-CdS(MMCC)光催化劑光催化產氫活性
界面處的差分電荷密度表明MoO2和Mo2C之間存在強烈的電子相互作用,電子從MoO2轉移到Mo2C,從而優化了電子結構和|ΔGH*|,促進了光生電荷的分離。MoO2的費米能級穿過導帶,驗證了其金屬特性。MoO2/Mo2C費米能級附近增強的局域態密度(圖5g)表明了本征電導率的提高,從而保證了快速電子轉移,有助于增強HER活性。MoO2/Mo2C異質結和MoO2的d帶中心分別位于-1.27eV和-1.15eV,表明在Mo2C形成后,d帶中心下移并遠離費米能級。因此,反鍵態被減少,助催化劑和吸附質之間的相互作用減弱,促進了H*從助催化劑表面的解吸,從而促進了H2的析出。
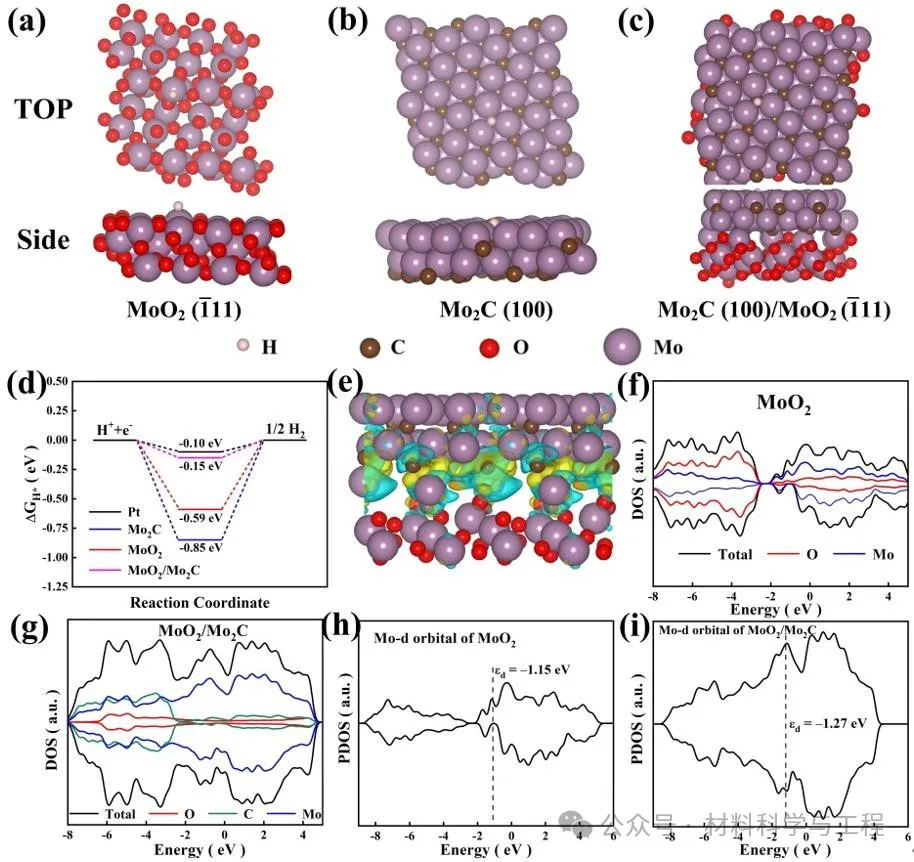
圖5 DFT理論計算
樣品的光電化學表征,證實了復合材料中增強的光吸收、電荷分離,這些因素共同作用提高光催化活性。
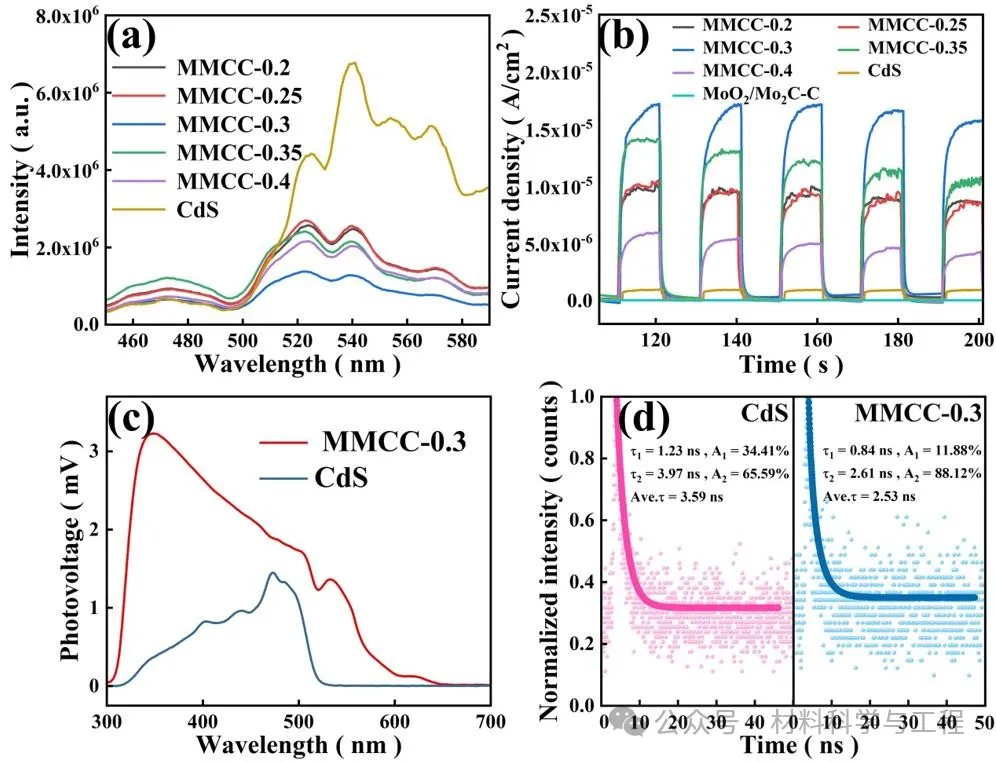
圖6光電化學表征
全文小結
綜上所述,作者成功構建了一種高活性、超穩定的MoO2/Mo2C-C助催化劑,用于CdS光催化析氫。雜化助催化劑中優化的氫結合能、d帶中心和強電子耦合賦予了MoO2/Mo2-C優異的助催化活性。值得注意的是,MoO2/Mo2C-CdS光催化劑具有出色的HER性能和耐用性。證明了優化的助催化劑、高導電碳橋、各種組分之間的高質量界面、增強的光吸收能力和抑制電荷復合的協同作用,從而獲得了優異的光催化性能。本文采用的策略為構建HER性能的其他有效助催化劑提供了有價值的見解。
通訊作者簡介
蘭亞乾:華南師范大學二級教授、博士生導師,教育部工程研究中心主任,英國皇家化學學會會士。中國化學會高級會員,中國化學會二氧化碳專業委員會副主任委員,中國化學會光化學專業委員會委員,中國感光學會光催化專業委員會副主任委員,中國化學快報(CCL)副主編,Natl. Sci. Rev.學科編輯組成員,Inorganic Chemistry、EnergyChem、Nano Research Energy、Polyoxometalates、物理化學學報、結構化學等期刊(顧問、青年)編委。主持國家自然科學基金杰出青年基金、優秀青年基金和面上項目等多項,近五年來以通訊作者在Nat. Synth.、Sci. Adv.、PNAS, Nat. Commun. (8)、J. Am. Chem. Soc. (13)、Angew. Chem. Int. Ed. (33)、Adv. Mater. (5)、Matter (2)、Chem (2)、Natl. Sci. Rev. (3)等期刊上發表通訊作者論文200余篇。論文被他引24000多次,ESI高引論文35篇,個人H-index 82。
李東升,博士/博士后,湖北省二級教授,博士生導師,教育部新世紀人才,享受國務院政府津貼專家、湖北省有突出貢獻中青年專家;湖北省自然科學創新群體負責人,國家級學科創新引智基地負責人,中國復合材料學會礦物復合材料專業委員會委員兼副秘書長,三峽大學副校長;主要從事無機化學、能源化學、材料化學方面的研究,主持國家自然科學基金面上項目(6項)、教育部重點科研基金、湖北省自然科學基金創新群體等項目20余項,在Acc. Chem. Res.、Coord. Chem. Rev.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、ACS Catal.等期刊發表論文180余篇,被引14000余次,H因子59;授權國家發明專利56件,出版著作3部,獲省部級科技一、二等獎6項; 入選愛思唯爾中國高被引學者和全球前2%頂尖科學家終身成就獎榜單。
第一作者簡介
喬秀清:副教授,碩士生導師,浙江大學博士,主要從事光催化材料研究。主持國家自然科學基金1項,以第一作者在Chemical Engineering Journal,J. Mater.Chem. A, Chinese Journal of Catalysis等期刊發表SCI收錄論文20余篇,論文被引1500余次,授權發明專利12項,指導研究生獲得三峽大學優秀碩士學位論文。主持湖北省一流線上、線下混合式課程《材料科學基礎》,指導學生多次獲得“互聯網+大學生創新創業大賽”湖北省金獎、銀獎及銅獎等。
免責聲明:本網站所轉載的文字、圖片與視頻資料版權歸原創作者所有,如果涉及侵權,請第一時間聯系本網刪除。
相關文章

官方微信
《腐蝕與防護網電子期刊》征訂啟事
- 投稿聯系:編輯部
- 電話:010-62316606
- 郵箱:fsfhzy666@163.com
- 腐蝕與防護網官方QQ群:140808414
點擊排行
PPT新聞

“海洋金屬”——鈦合金在艦船的
點擊數:9037

腐蝕與“海上絲綢之路”
點擊數:7224